- 移动端


武汉枢密脑科学技术有限公司品牌商
8 年
手机商铺
- NaN
- 0.7000000000000002
- 1.7000000000000002
- 0.7000000000000002
- 3.7

武汉枢密脑科学技术有限公司
入驻年限:8 年
- 联系人:
枢先生
- 所在地区:
湖北 武汉市 江夏区
- 业务范围:
抗体、试剂、细胞库 / 细胞培养、技术服务、耗材、实验室仪器 / 设备
- 经营模式:
生产厂商 经销商
推荐产品


公司新闻/正文
【客户文章-AAV在肾脏疾病中的研究应用】东南大学刘必成/王彬教授团队揭示27-HC代谢失调增强缺血性AKI中近端肾小管上皮细胞铁死亡敏感性
27 人阅读发布时间:2026-06-16 10:17
急性肾损伤(AKI)是临床常见的危重症,肾小管上皮细胞的缺血性损伤被广泛认为是AKI的主要起始事件。在缺血性AKI过程中,肾小管细胞死亡是关键的早期事件,且伴随大量能量需求。尤其是位于皮髓质区的近端肾小管细胞,因其高代谢需求和有限的氧供,对缺氧特别敏感,易于发生广泛的细胞死亡。近年来,铁死亡(ferroptosis)——一种铁依赖性的、以脂质过氧化和细胞膜破裂为特征的调节性细胞死亡方式——已被证实在缺血性AKI中发挥重要作用。然而,连接肾小管脂质代谢失调与铁死亡之间的确切机制仍不明确,这构成了当前AKI发病机制理解中的关键空白。
近端小管(PT)细胞中的胆固醇失调与肾脂毒性有关。研究表明,AKI后血清胆固醇和甘油三酯升高,其氧化产物可加剧局部炎症反应并导致细胞损伤。值得注意的是,氧化甾醇作为胆固醇的早期氧化中间产物,在非肾脏组织中被发现具有免疫调节或细胞毒性作用,但氧化甾醇在肾脏铁死亡中的具体贡献,尤其是缺血性损伤背景下,尚待充分阐明。
来自东南大学附属中大医院刘必成教授和王彬教授团队在Journal of the American Society of Nephrology(JASN)期刊上发表了题为“Metabolic Dysregulation of 27-Hydroxycholesterol Sensitizes Proximal Tubular Epithelial Cells to Ferroptosis in Ischemic Acute Kidney Injury”的研究成果,创新性地采用单细胞RNA测序、空间转录组学与靶向代谢组学等多组学整合策略,系统解析了近端直小管(PST)细胞在肾脏缺血再灌注损伤(IRI)早期的时空动态代谢重塑规律。研究团队发现,缺血导致降解27-羟基胆固醇(27-HC)的关键酶CYP7B1显著下调,致使27-HC在PST中特异性蓄积。在分子层面上,蓄积的27-HC作为内源性配体直接激活雌激素受体α(ERα),引发下游促铁死亡蛋白血红素加氧酶1(HMOX1)的异常激活,进而增加肾PST细胞对铁死亡的敏感性,阻碍其正常修复。利用腺相关病毒(AAV)在小鼠肾脏过表达Cyp7b1可有效促进27-HC降解,全面减轻缺血性AKI的进展。该发现首次揭示了CYP7B1/27-HC代谢轴在调节肾脏铁死亡中的关键作用,为阻断AKI向慢性肾脏病(CKD)转变提供了全新的转化医学靶点。

1、外髓外带的PST对肾缺血损伤最为敏感
为明确AKI向CKD进展的空间异质性特征,研究团队对小鼠单侧缺血再灌注(UIR)模型不同时间点的肾脏组织开展空间转录组分析,并结合既往单细胞测序数据解析肾脏分区。无监督聚类将肾脏划分为皮质、外髓外带、外髓内带、内髓四个区域,其中基于炎症和纤维化相关基因表达计算的损伤评分显示,外髓外带在UIR后所有时间点的损伤程度均最高,肾损伤标志基因Havcr1在此区域特异性高表达,与该区域的缺血易感性完全匹配。空间转录组进一步证实,外髓外带主要由PST细胞构成,损伤后该细胞特征性标志基因Slc27a2的表达显著降低,提示细胞发生去分化损伤。
研究同时纳入健康人、AKI及CKD患者的肾活检样本进行免疫荧光验证,结果显示SLC27A2在AKI早期与肾损伤标志物VCAM1共定位,在CKD晚期则与纤维化标志物COL1A1共定位。上述结果明确了AKI向CKD进展的区域动态变化,证实外髓外带的PST细胞是缺血性肾损伤的核心易感细胞群体,为后续机制研究锁定了靶细胞类型。
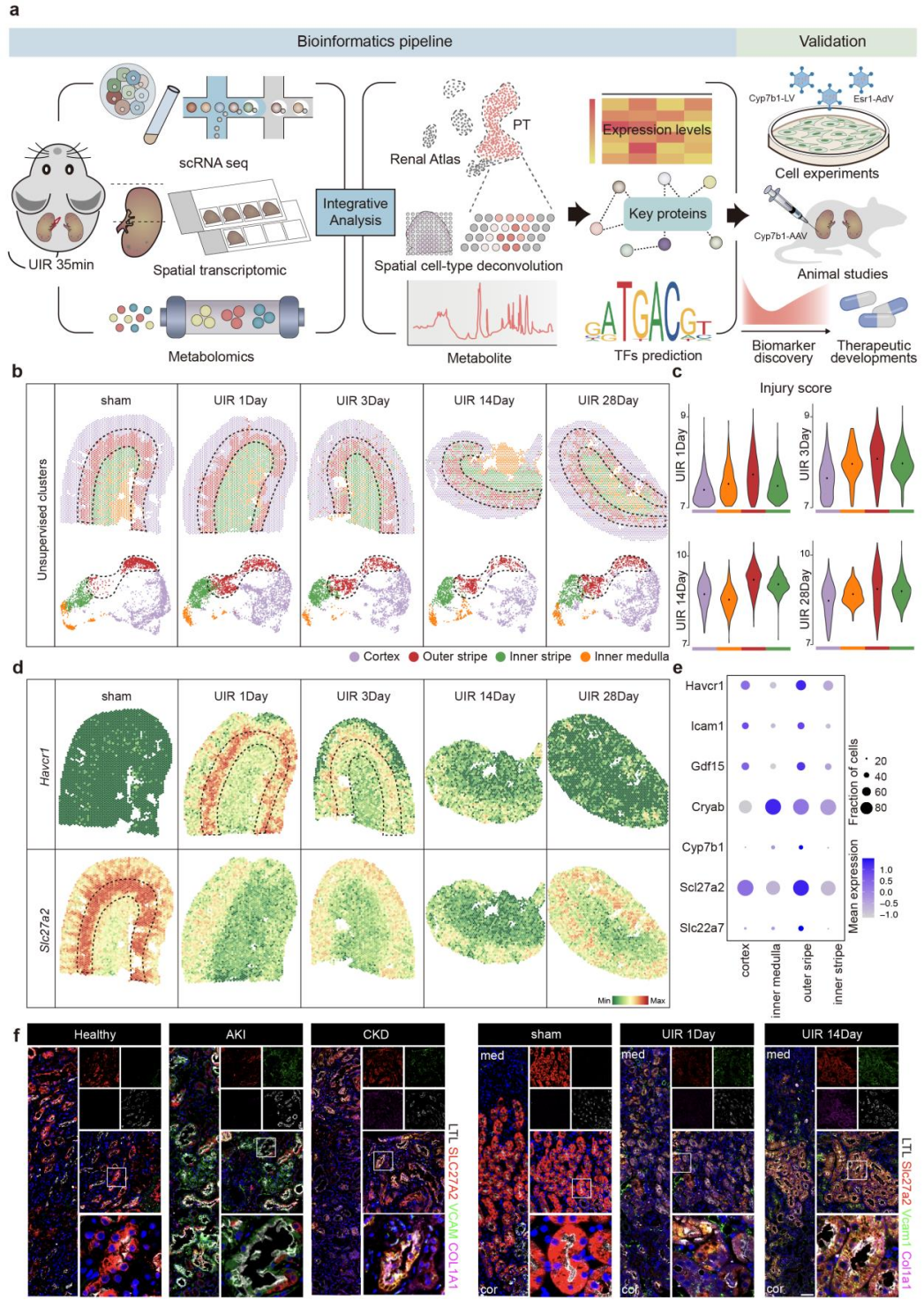
图1. UIR后近端肾小管细胞损伤的时空动态变化
2、损伤后的PST细胞在UIR后对铁死亡高度敏感
研究团队进一步对20余万PT细胞进行聚类,并从中聚焦PST细胞,将其进一步划分为9个亚群。其中PST-5亚群在UIR后第1天占比从5%急剧升至17%,随后持续下降,符合“修复失败型PT”的表型特征,且持续高表达Havcr1、Vcam1等损伤标志基因。细胞死亡通路评分显示,铁死亡是AKI向CKD进展中PT细胞最主要的死亡方式,而PST-5的铁死亡评分显著高于其他亚群。与之相印证,铁死亡关键调控蛋白ACSL4在PST-5中呈特异性高表达。
免疫荧光证实,在AKI患者及小鼠肾脏中,VCAM1、SLC27A2与ACSL4共定位于PST细胞,且三者表达水平与血清肌酐呈正相关。动物实验显示,单侧及双侧缺血再灌注(uIRI、bIRI)小鼠肾脏中,肾小管损伤标志物VCAM1、KIM1上调,铁死亡抑制蛋白GPX4下调,且这些变化具有肾小管细胞特异性。以上结果证实,修复失败型PST细胞在缺血损伤后对铁死亡高度敏感。
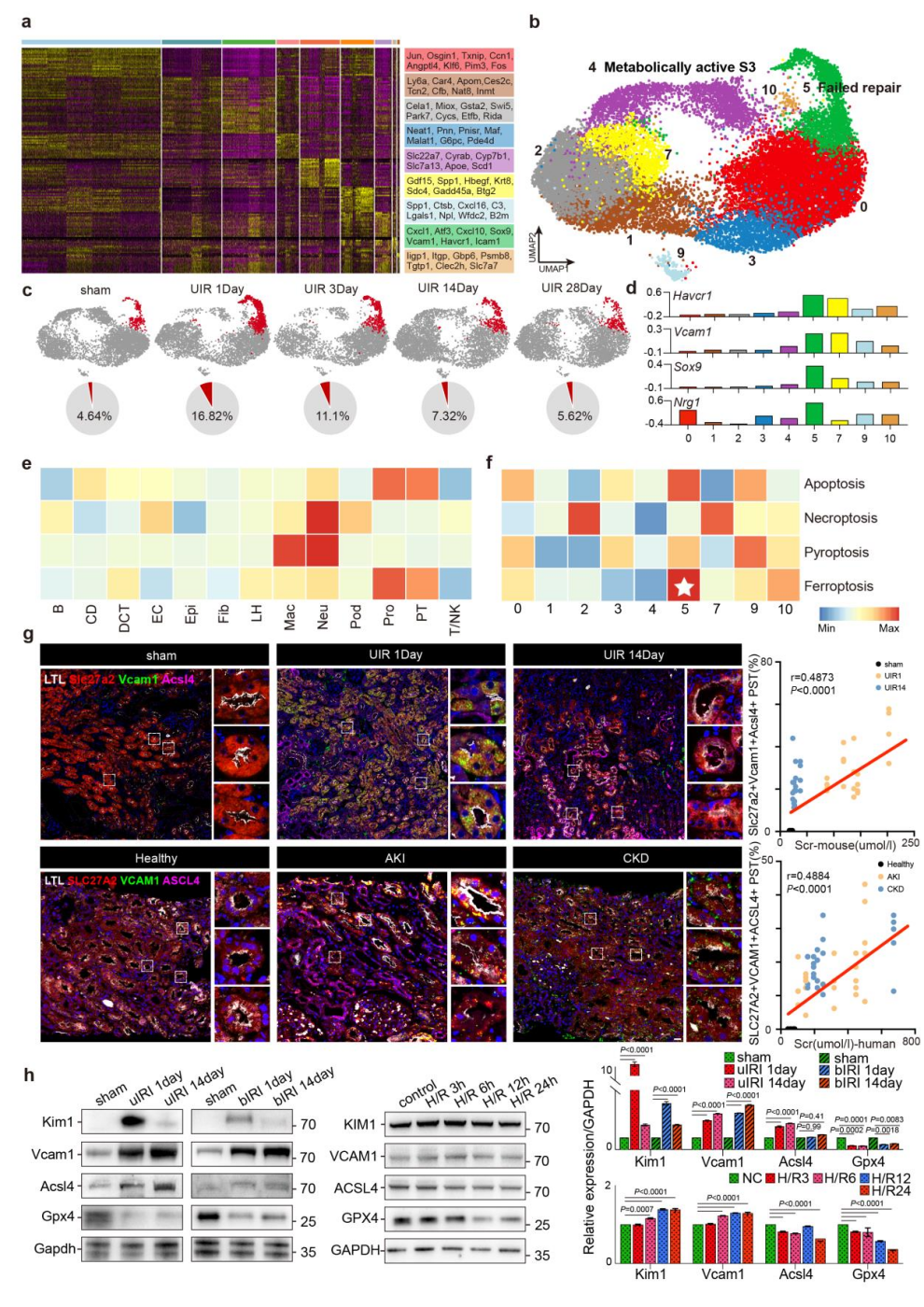
图2. UIR后PST细胞对铁死亡的高敏感性
3、受损的PST细胞在AKI后下调CYP7B1表达以富集27-HC
团队进一步探究了修复失败型PST细胞形成的代谢驱动因素。RNA速率分析显示PST-4是PST-5的前体细胞,基因本体(GO)富集分析表明PST-4亚群的胆固醇代谢通路显著活化。靶向代谢组学发现,小鼠肾脏中27-HC在缺血后第1天急剧升高,第3天逐渐下降,慢性期恢复正常,这一趋势与PST细胞的动态变化相吻合。
在胆固醇代谢通路中,CYP27A1催化胆固醇生成27-HC,而CYP7B1负责将27-HC进一步代谢清除。单细胞测序显示,缺血早期Cyp27a1表达无显著变化,而Cyp7b1表达显著下调,且CYP7B1是PST-4亚群的特异性标志物。空间转录组、蛋白质印迹及免疫荧光实验进一步验证,CYP7B1在uIRI、bIRI小鼠AKI早期肾脏组织及缺氧处理的HK2细胞(人近端肾小管上皮细胞系)中均显著降低,AKI后慢性晚期Cyp7b1表达持续处于低水平,而Vcam1表达升高。以上结果表明,PST细胞通过下调Cyp7b1促进27-HC异常蓄积,该过程在缺血性AKI早期尤为关键。
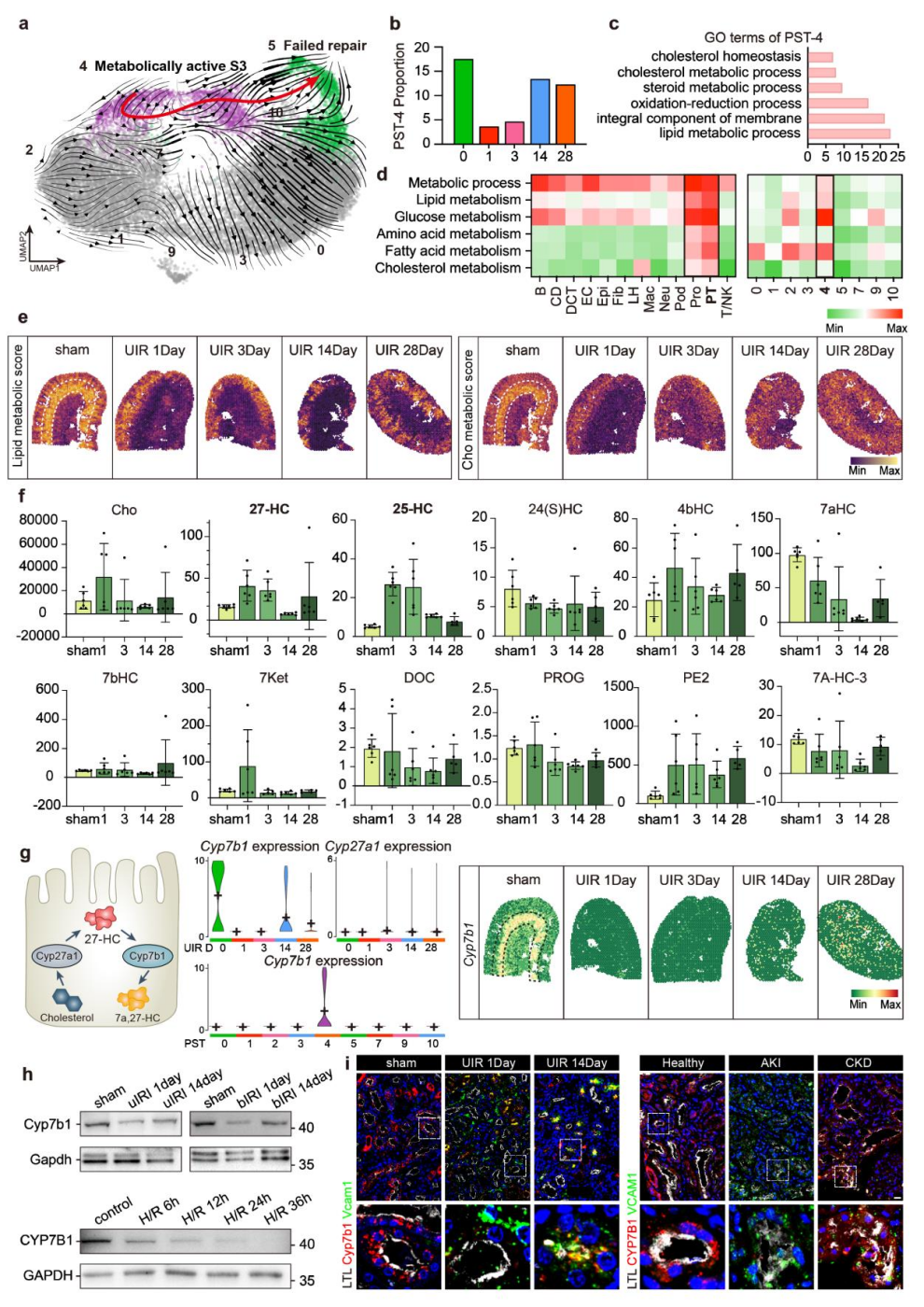
图3. PST细胞中CYP7B1缺失导致27-HC蓄积
4、27-HC增强PST细胞对铁死亡的敏感性
体外实验表明,27-HC处理HK2细胞可剂量依赖性降低细胞活力,下调GPX4和CYP7B1蛋白水平,同时上调促铁死亡酶HMOX1及27-HC的天然配体ERα,并诱导脂质过氧化和胞内脂质蓄积,证实27-HC可直接增强细胞对铁死亡的敏感性。进一步构建稳定过表达CYP7B1的HK2细胞系,结果显示CYP7B1基因过表达可显著降低细胞27-HC水平,减少ACSL4表达和脂质过氧化,恢复GPX4水平,表明CYP7B1过表达可抑制铁死亡。
机制研究显示,27-HC与雌二醇(E2)同为ERα配体,但二者结合ERα后对下游HMOX1的调控效应截然相反:27-HC上调HMOX1表达,而E2则抑制HMOX1。蛋白互作分析和免疫共沉淀证实ERα与HMOX1存在直接相互作用。此外,转录因子Atf3可结合Cyp7b1启动子并抑制其转录;敲低Atf3可恢复Cyp7b1水平,下调Acsl4和Hmox1,减轻缺氧复氧诱导的铁死亡。综上,27-HC是强效的铁死亡诱导剂,其胞内丰度受CYP7B1调控,并通过ERα-HMOX1通路决定缺血后PST细胞的铁死亡敏感性。
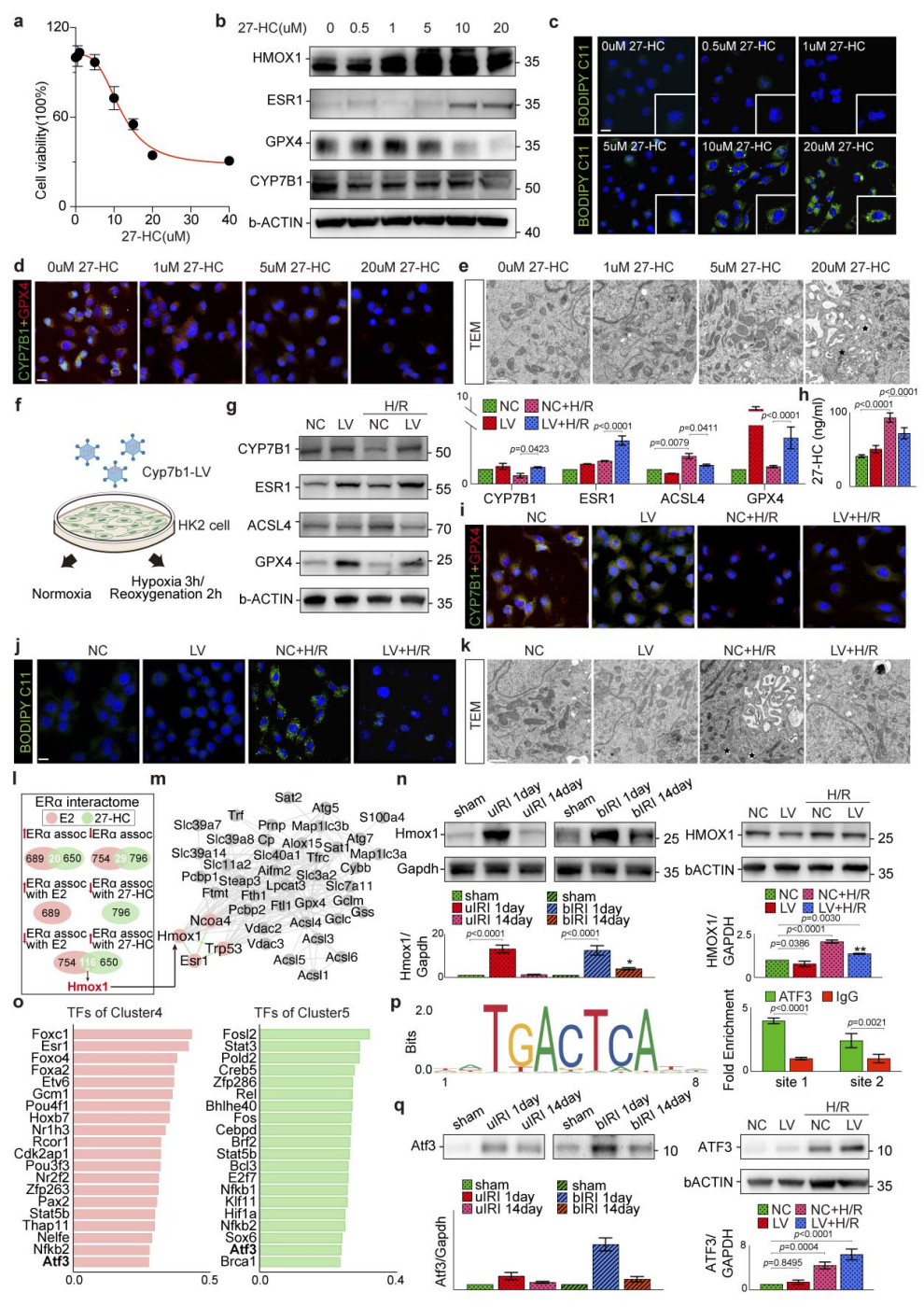
图4. 27-HC增强PST细胞对铁死亡的敏感性
5、体内过表达Cyp7b1通过下调27-HC减轻肾脏IRI损伤
为验证CYP7B1/27-HC轴的体内治疗潜力,研究团队通过肾实质原位注射AAV载体,在小鼠肾脏实现Cyp7b1特异性过表达。结果显示,Cyp7b1过表达可显著降低肾脏组织27-HC水平,有效改善IRI小鼠的肾功能(血清肌酐、血尿素氮水平显著降低),同时减轻了肾组织病理损伤和纤维化程度。透射电镜及分子检测证实,Cyp7b1过表达可显著抑制肾组织中的铁死亡,表现为胞内脂质蓄积减少、Acsl4和Hmox1下调、Gpx4上调。上述动物实验结果证实,体内过表达Cyp7b1可通过降解27-HC、抑制铁死亡,发挥显著的肾保护作用,提示CYP7B1/27-HC轴可作为缺血性AKI的潜在干预靶点。
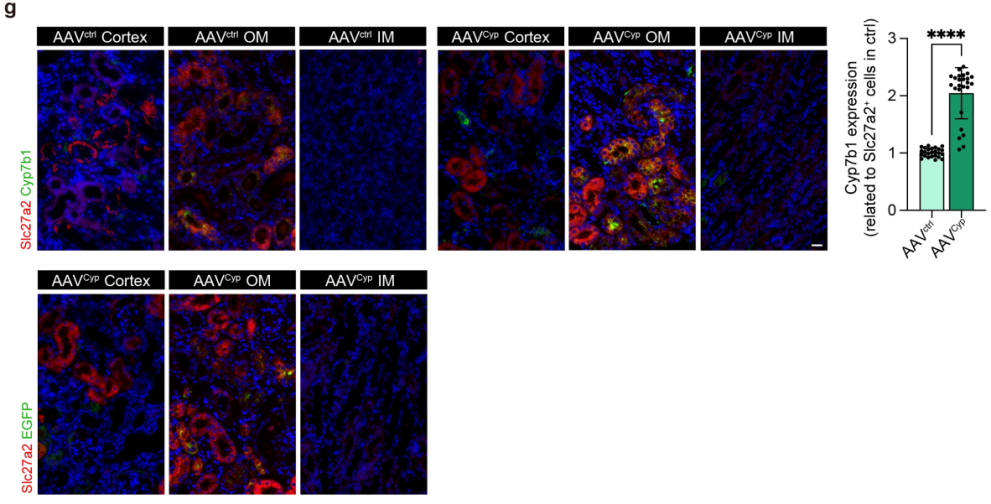
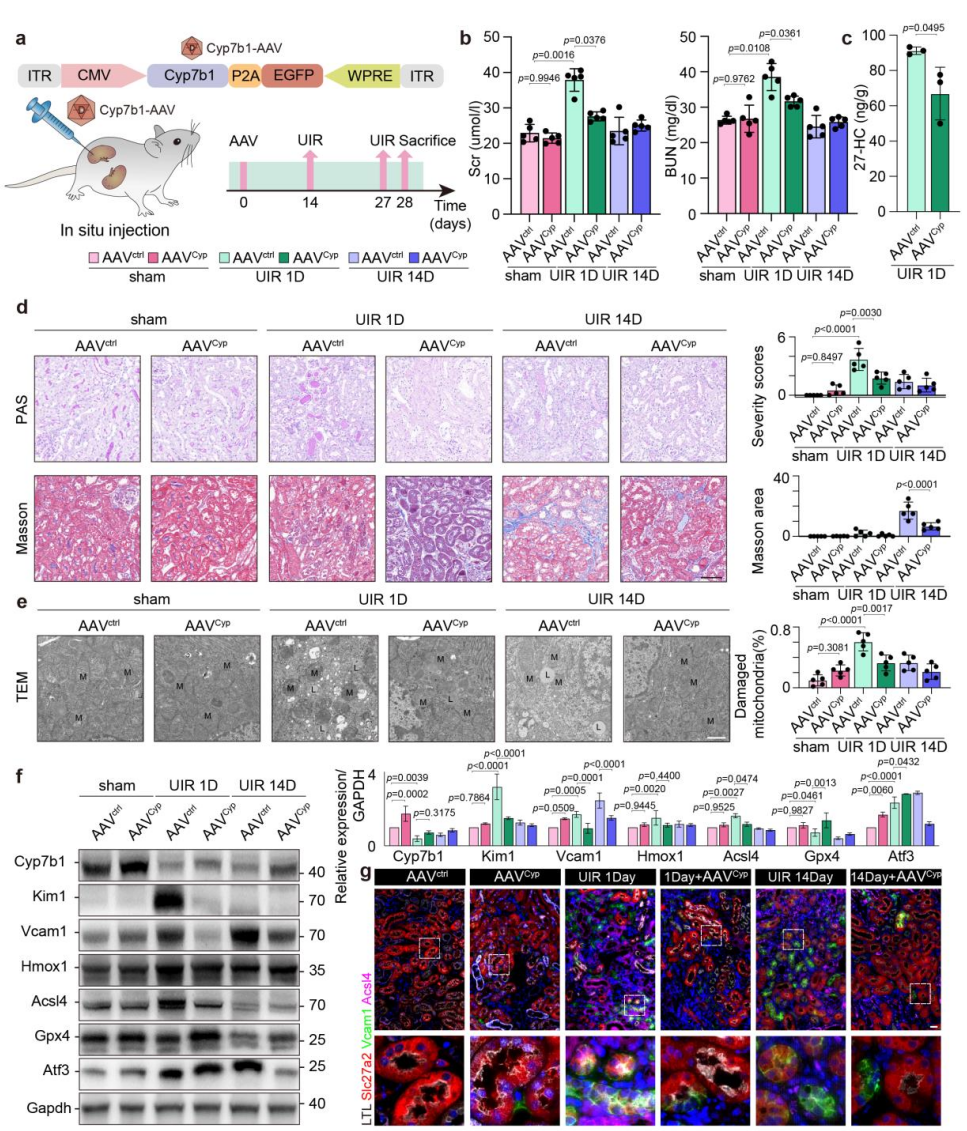
图5. 体内过表达Cyp7b1减轻IRI损伤
✦ 总结
该研究依托多组学联合体内外功能实验,首次阐明CYP7B1/27-HC/ERα/HMOX1信号轴调控缺血性AKI PST铁死亡的新机制:缺血损伤造成PST细胞Cyp7b1表达下调,促使27-HC大量蓄积;27-HC作为内源性配体结合ERα并上调Hmox1,通过扰乱铁稳态、加重脂质过氧化增强细胞铁死亡敏感性,造成肾小管修复障碍,驱动AKI向CKD演进。研究首次建立了胆固醇代谢紊乱与肾铁死亡的直接因果关联,拓宽了缺血肾损伤的研究思路。当然研究也存在一定局限:尚未利用基因敲除模型验证内源Cyp7b1生理作用,AAV在肾脏不同细胞类型中的转染模式需细化。后续可围绕该通路开发CYP7B1激动剂、27-HC或HMOX1靶向药物,助力缺血性AKI新药研发。
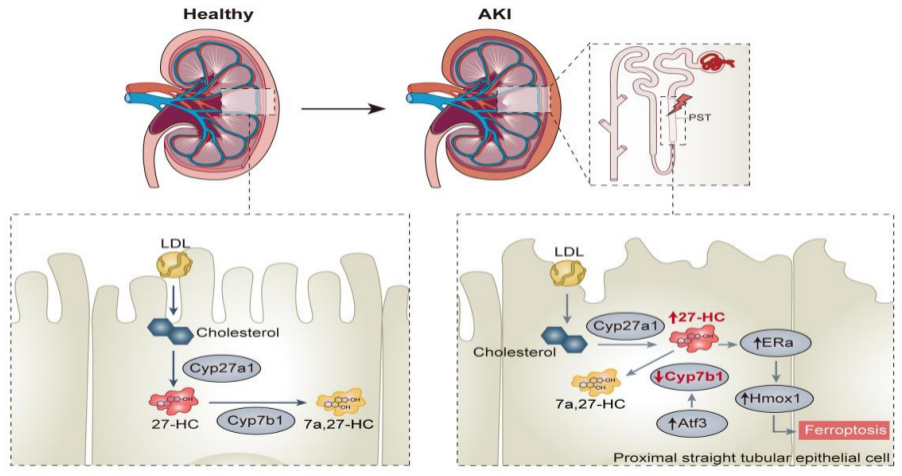
图6. CYP7B1/27-HC轴调控PST细胞铁死亡敏感性模式图
本文使用的来自枢密科技的病毒产品,列表如下:

如有相关需求,或了解更多产品服务,欢迎咨询我们!点击进入店铺,查看更多产品及服务