- 移动端


武汉枢密脑科学技术有限公司品牌商
8 年
手机商铺
- NaN
- 0.7000000000000002
- 1.7000000000000002
- 0.7000000000000002
- 3.7

武汉枢密脑科学技术有限公司
入驻年限:8 年
- 联系人:
枢先生
- 所在地区:
湖北 武汉市 江夏区
- 业务范围:
抗体、试剂、细胞库 / 细胞培养、技术服务、耗材、实验室仪器 / 设备
- 经营模式:
生产厂商 经销商
推荐产品


公司新闻/正文
IF=22.9【AAV在心脏疾病中的应用】USP18“刹车”线粒体自噬,PTEN‑L是“帮凶”——武汉大学中南医院团队揭示心肌I/R损伤新靶点
37 人阅读发布时间:2026-06-15 11:32
急性心肌梗死(AMI)由冠状动脉突发阻塞引发心肌缺血所致,若未及时恢复冠脉灌注,会进展为心肌瘢痕化与不良心脏重构,最终诱发心力衰竭。经皮冠状动脉介入治疗(PCI)是AMI的初始治疗,可快速重建冠脉血流,但再灌注过程会诱发一系列细胞损伤,甚至可能抵消血运重建的获益,目前临床仍无有效手段防治缺血/再灌注(I/R)损伤。深入研究再灌注损伤机制可为AMI研发新疗法,助力提升病患预后。
维持线粒体稳态是细胞生理功能正常运转的关键,线粒体自噬作为清除受损、异常或功能失调线粒体的核心机制,其功能异常与心肌I/R损伤的发生发展密切相关。恩格列净(SGLT2抑制剂)通过激活线粒体自噬,对心脏微血管I/R损伤提供保护作用。因此,靶向线粒体稳态与线粒体自噬已成为干预心肌I/R损伤的潜在方向。PINK1(PTEN诱导激酶1)-Parkin轴是经典的线粒体自噬调控通路,线粒体去极化促使PINK1在线粒体外膜蓄积、募集Parkin,后者泛素化线粒体外膜蛋白,最终启动线粒体自噬清除。研究表明,PINK1-Parkin通路在心肌I/R损伤中发挥保护作用。磷酸酶和张力蛋白同源物长亚型(PTEN-L)通过蛋白磷酸酶活性使泛素结合物去磷酸化发挥其作用,被证实是Parkin介导线粒体自噬的新型负向调控因子。因此,靶向PTEN-L-Parkin-线粒体自噬通路具备治疗心肌I/R损伤的潜力。
泛素化与去泛素化修饰在线粒体稳态调控中占据核心地位,尤其是在心血管疾病和心肌I/R损伤等病理条件下。多种去泛素化酶,包括泛素特异性蛋白酶(USP)30、USP33和USP35,已被证实参与心肌细胞线粒体自噬的调控。USP18既往研究主要聚焦于其在炎症、抗病毒信号及肿瘤进展中的作用,近期研究发现其可缓解主动脉缩窄诱导的病理性心脏重构,提示该分子可能参与心肌I/R损伤的调控,但USP18在心肌I/R损伤中的具体功能,以及其调控线粒体自噬的分子机制尚未明确。
基于上述临床问题与研究空白,本研究旨在明确USP18是否通过调控线粒体自噬参与心肌I/R损伤过程,并深入解析其潜在分子机制,以期为心肌I/R损伤的防治提供新的潜在治疗靶点。2026年3月24日,武汉大学中南医院心血管内科主任医师鲁志兵、胡笑容在Military Medical Research(IF=22.9)发表题为“USP18 exacerbates myocardial I/R injury by inhibiting Parkin mitophagy through the deubiquitinase PTEN-L”研究论文。USP18在I/R损伤后的小鼠心脏组织及缺血性心脏病(IHD)患者心脏组织中表达均上调。心肌特异性USP18缺失可缓解I/R诱导的急性心肌损伤、线粒体功能障碍及不良心脏重构,而USP18过表达则会加重上述病理改变。机制上,USP18与PTEN-L结合,PTEN-L进一步结合Parkin并抑制其磷酸化及线粒体转位,最终抑制线粒体自噬。Parkin敲低可消除USP18缺失带来的心脏保护作用,而PTEN-L敲低则可逆转USP18过表达的损伤效应。此外,PTEN-L还可通过旁分泌机制发挥致病作用,使用中和抗体阻断PTEN-L可减轻心肌I/R损伤。ST段抬高型心肌梗死(STEMI)患者(尤其是介入治疗后)血清PTEN-L水平显著升高。综上所述,USP18通过PTEN-L-Parkin轴抑制线粒体自噬,并加重心肌I/R损伤,该机制涉及细胞内和旁分泌途径。靶向USP18-PTEN-L通路有望成为减轻心肌I/R损伤的新型治疗策略。

1.心肌I/R损伤可诱导USP18表达
为探寻心肌I/R损伤的关键调控分子,研究对I/R处理(左前降支(LAD)缺血45分钟后再灌注)24小时的小鼠左心室(LV)组织进行基因表达谱分析,GO功能富集分析提示差异基因显著富集于泛素介导的蛋白调控通路;进一步对泛素相关差异表达基因筛选后,共鉴定出14个上调基因与2个下调基因,其中USP18在I/R心脏中的上调趋势最为显著。临床样本检测发现,IHD患者病变心肌组织中USP18蛋白表达明显升高;重新分析终末期IHD患者心脏RNA测序数据集GSE203160,也印证了这一上调趋势。动物实验表明,小鼠心肌I/R损伤后24小时至4周内,USP18表达逐渐增加。USP18与小麦胚芽凝集素(WGA标记心肌细胞膜)的双重免疫荧光染色显示,正常心肌组织中USP18本底表达极低,I/R损伤后表达显著升高,且主要定位于心肌细胞,而心肌成纤维细胞、内皮细胞及巨噬细胞中的USP18表达水平,在I/R应激下未见变化。
体外原代新生大鼠心室肌细胞(NRVM)实验同样证实,缺氧/复氧(H/R,细胞在95% N₂、5% CO₂中缺氧4小时,再换至95%空气、5% CO₂中复氧过夜)损伤可在6、12、24小时时间点显著诱导USP18表达,且该蛋白主要定位于细胞质。综上,体内外结果一致表明心肌I/R损伤可诱导USP18表达。
进一步探究USP18的表达调控机制发现,在NRVM的H/R模型中,TNF-α及心肌梗死关键炎症介质HMGB1(高迁移率族蛋白)可通过转录水平正向调控USP18表达,是介导急性心肌I/R损伤诱导USP18上调的重要上游炎症因子。
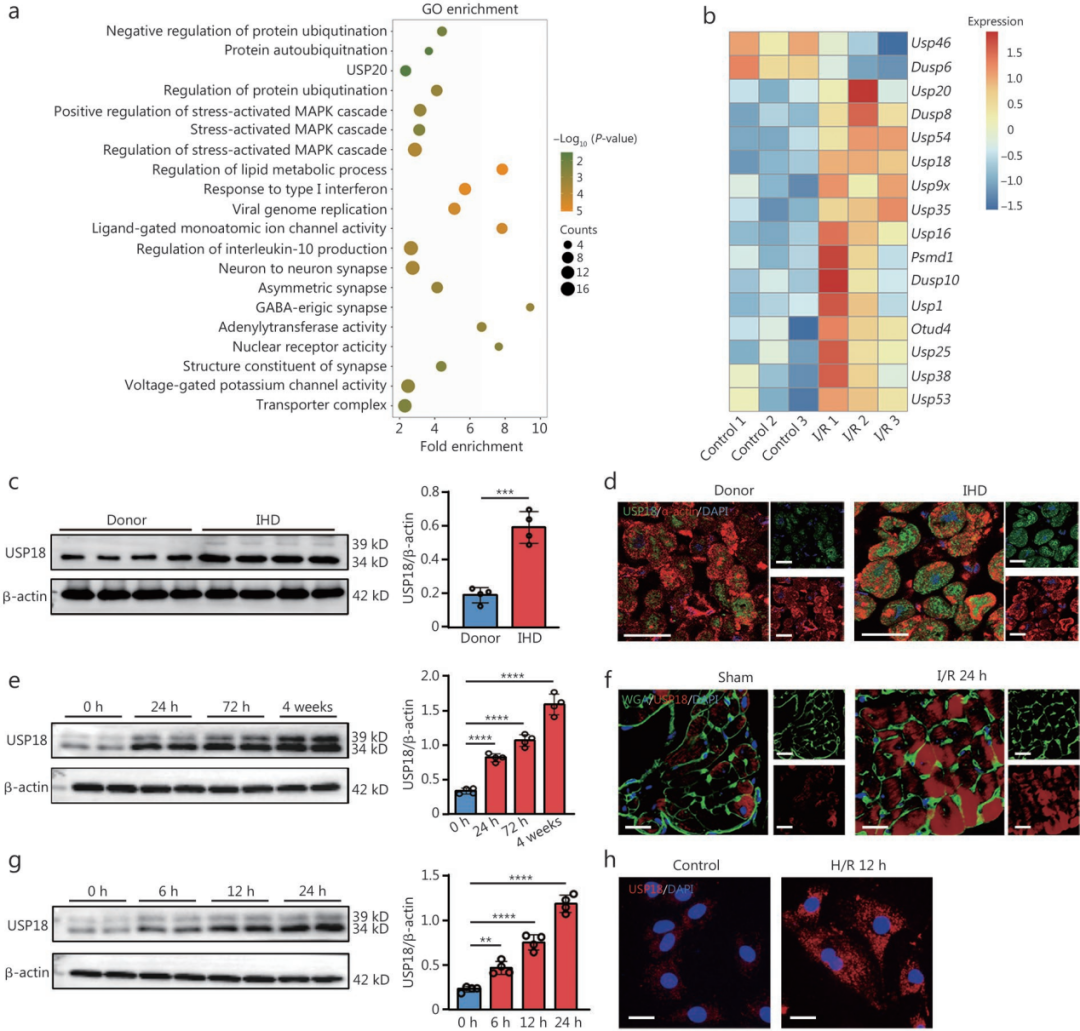
图1.心肌I/R损伤可诱导USP18表达
2.USP18缺失减轻小鼠体内线粒体功能障碍、急性心肌I/R损伤及心脏重构
为明确USP18在心肌I/R损伤中的作用,作者构建了心肌特异性USP18条件性敲除(USP18-cKO)小鼠。结果发现,与野生型(WT)相比,USP18-cKO小鼠在相同手术处理后,I/R损伤后的梗死面积更小;术后4小时血清cT-nT、CK-MB和LDH水平均显著降低,表明心肌损伤得到缓解。USP18缺失使LV组织DNA碎片化减少、cleaved caspase-3活性下降,提示I/R损伤后细胞总凋亡减少。
随后评估线粒体形态和功能。结果显示,USP18-cKO小鼠表现出线粒体DNA含量增加,线粒体复合物Ⅰ和Ⅱ-Ⅲ活性改善;I/R诱导的线粒体丢失和嵴排列紊乱得到缓解,线粒体结构得以维持。此外,USP18敲除也减轻了I/R诱导的线粒体功能障碍,表现为耗氧率(OCR)升高,基础呼吸、ATP相关呼吸和最大呼吸水平均升高,而备用呼吸无显著差异。
为进一步探究USP18对生理条件下心功能的长期影响及对I/R后心脏重构的作用,作者延长了观察时间。USP18-cKO小鼠在2、4、6和12月龄时心功能与WT同窝对照相当。与WT相比,USP18-cKO小鼠心肌细胞增大程度更轻,LV纤维化重构减轻,心重/胫骨长比(HW/TL)降低。I/R后4周,USP18-cKO心脏中肥大和纤维化标志物(包括心房钠尿肽(ANP)、B型钠尿肽(BNP)以及胶原Ⅰ和Ⅲ)的转录激活被显著抑制。I/R后USP18-cKO小鼠心功能改善、心室扩张减轻,表现为左心室射血分数(LVEF)和左心室短轴缩短率(LVFS)升高,同时舒张期左心室内径(LVIDd)、收缩期左心室内径(LVIDs)及心率降低。总体而言,心肌特异性敲除USP18揭示了USP18在小鼠心脏中对线粒体破坏、急性I/R诱导的心肌损伤及长期不良心室重构具有致病性贡献。
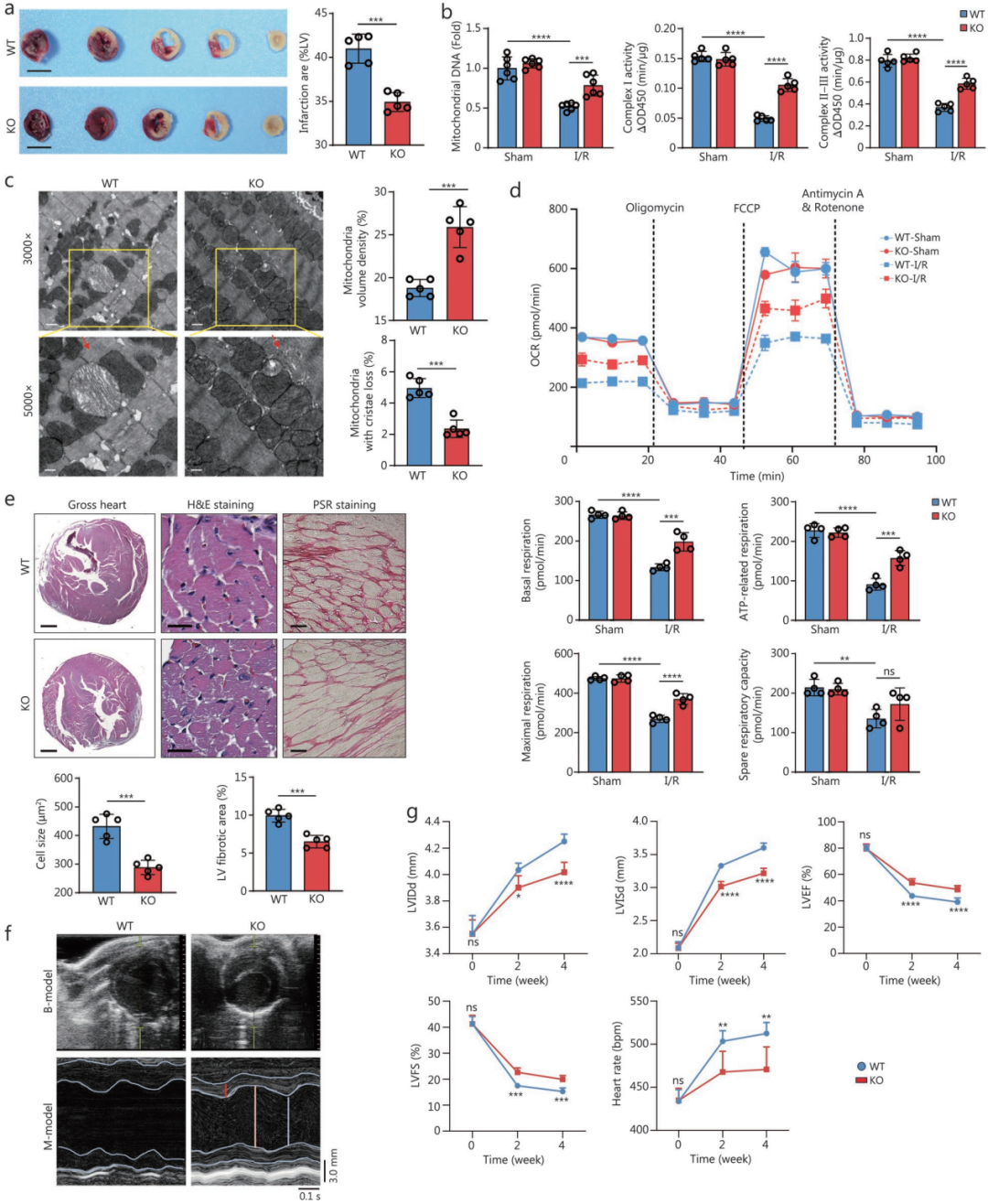
图2.USP18缺失可减轻小鼠体内线粒体功能障碍、急性心脏I/R损伤及心脏重构
3.心肌USP18过表达对线粒体功能及缺血后重构的病理性影响
为探究USP18过表达是否会加重上述病理改变,作者在I/R术前两周经尾静脉向C57BL小鼠单次注射AAV9-USP18,使其心脏组织中USP18呈高表达。USP18过表达(USP18-OV)小鼠在I/R术后24小时梗死面积增大、LV组织DNA碎片增加,术后4小时血清cTnI、CK-MB和LDH水平升高。此外,心肌过表达USP18显著加重了线粒体功能障碍:I/R小鼠线粒体密度降低、嵴排列紊乱加重、线粒体DNA含量减少,复合物Ⅰ和Ⅱ-Ⅲ活性下降;同时细胞OCR降低,基础、最大和备用呼吸均受抑制。
生理状态下,USP18-OV小鼠从6月龄起心功能下降,至12月龄时更为明显。I/R术后4周,USP18-OV小鼠出现明显的心脏重构,表现为心肌细胞肥大、纤维化区域扩大、HW/TL升高,心肌肥厚与纤维化标志物的mRNA转录水平也升高;超声提示LVEF和LVFS降低,LVIDd、LVIDs及心率均恶化。总之,这些发现表明,心肌特异性过表达USP18会加重I/R后心脏的线粒体功能障碍、急性缺血性损伤以及后续的结构和功能恶化。
体外H/R模型进一步证实,采用腺病毒过表达USP18可加重心肌细胞损伤及线粒体功能障碍,而采用siRNA沉默USP18则减轻上述改变。
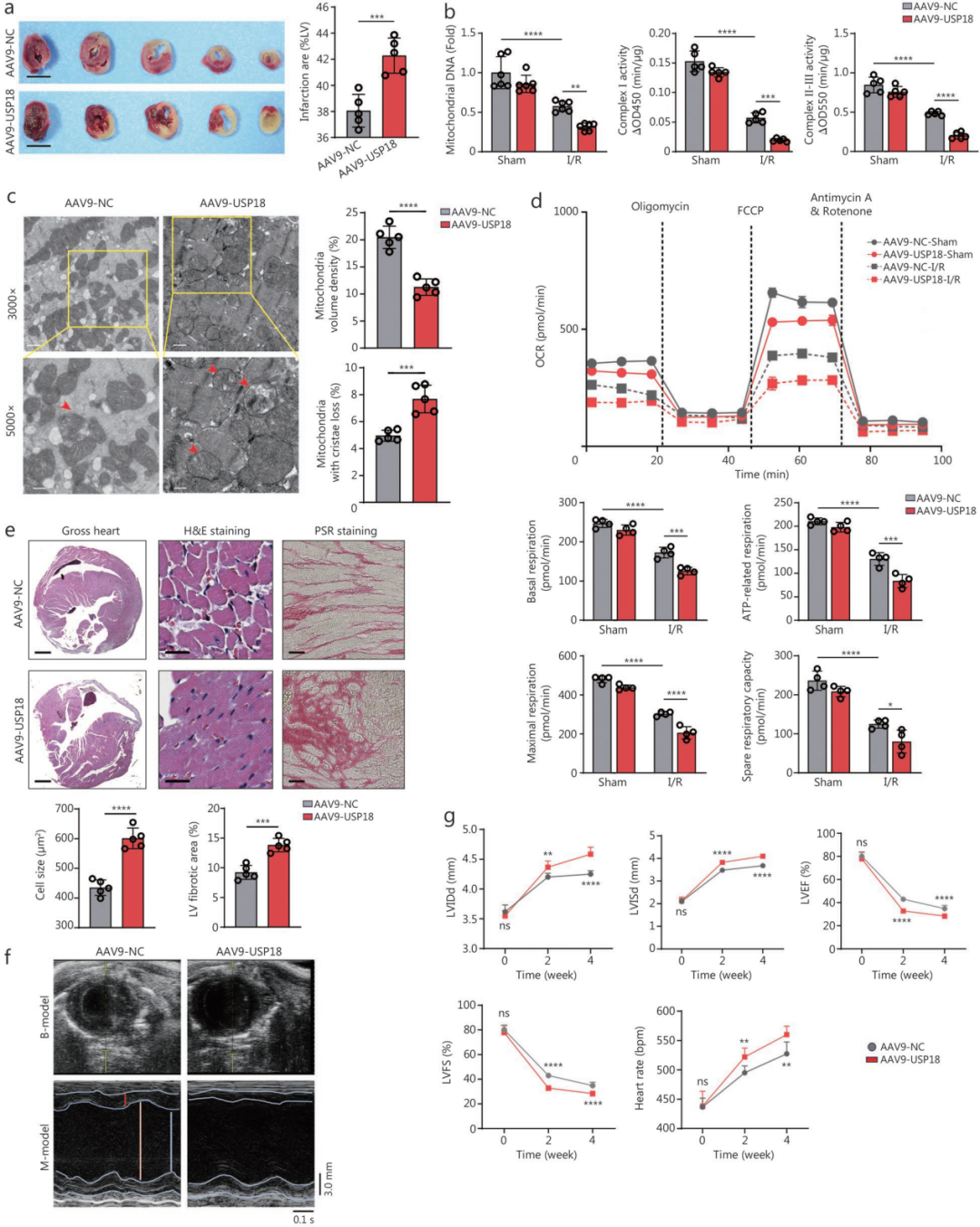
图3.心肌过表达USP18会加重小鼠I/R后的线粒体功能障碍、急性心肌损伤及心脏重构
4.USP18通过调控线粒体功能并抑制线粒体自噬,加剧心肌I/R损伤
为明确USP18的作用,研究者对I/R后24小时的USP18-cKO和WT小鼠心脏进行了RNA-seq。KEGG分析显示,I/R后USP18缺失心脏中最显著富集的通路是代谢调控和线粒体自噬;热图表明USP18-cKO显著调控了线粒体活性和代谢相关基因的表达。GSEA进一步证实,USP18缺失显著上调了线粒体相关的GO通路。
敲除和过表达实验均证实USP18影响线粒体功能。线粒体功能障碍是心肌I/R损伤的核心环节,而线粒体自噬受损会加速这一过程,提示USP18可能通过调控线粒体自噬参与I/R损伤。
透射电镜显示,I/R小鼠心脏中线粒体自噬受损,而在USP18-cKO心脏中得到恢复。I/R损伤后,线粒体中PINK1、Parkin、P62、LC3Ⅱ和泛素化蛋白(Ub)水平升高;与WT相比,USP18-cKO小鼠上述蛋白水平更高,而USP18-OV小鼠则更低。
体外实验用线粒体自噬和溶酶体双荧光染料检测发现:红色标记自噬线粒体、绿色标记溶酶体;当自噬体与溶酶体融合后,进入酸性环境的红色荧光增强,反映自噬流活性。结果显示,敲低USP18后自噬流增强,过表达则减弱。USP18敲低使NRVM线粒体中PINK1、Parkin、Ub、P62和LC3Ⅱ水平升高,过表达则降低。进一步证实USP18通过抑制PINK1/Parkin介导的线粒体自噬,在I/R损伤中发挥有害作用。
此外,USP18敲除或过表达并不改变线粒体裂变(p-Drp1)、融合(MFN1/2)、生物发生(PGC-1α、TFAM)及其他自噬受体(BNIP3、FUNDC1)的水平,说明USP18主要选择性抑制PINK1/Parkin通路,而非全面破坏线粒体质量控制。与此一致,USP18可上调凋亡蛋白Bax/Bcl-2,这一效应在敲除后被抑制。同时,H/R应激下敲低USP18可降低线粒体活性氧(ROS)水平,过表达则升高(线粒体清除增强可能是ROS减少的原因)。此外,USP18-cKO小鼠I/R后促炎因子(TNF-α、IL-1、IL-6)水平也降低。
综上,USP18主要通过抑制线粒体自噬来影响线粒体功能,进而调控健康线粒体数量、呼吸功能、氧化应激和凋亡。
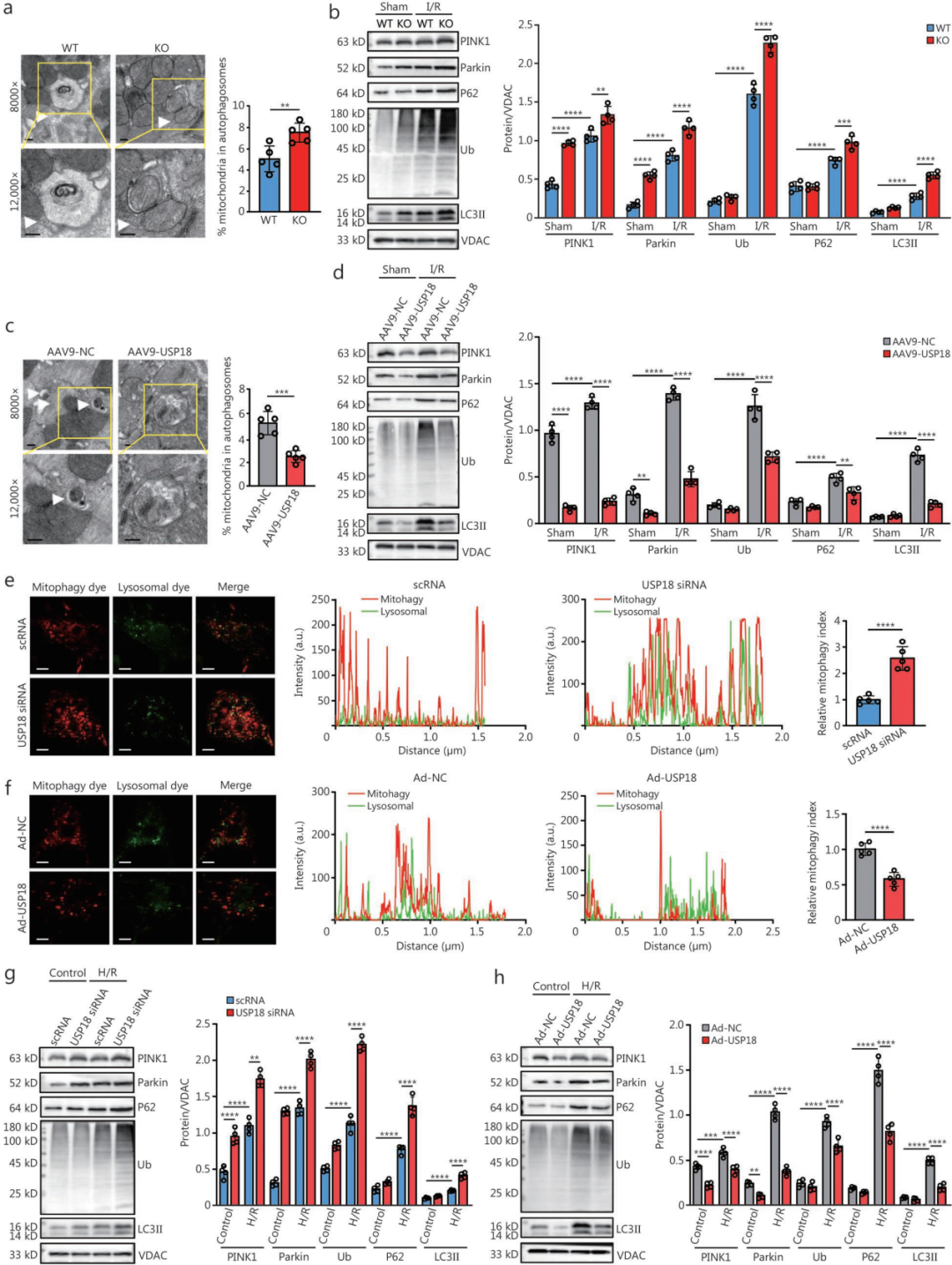
图4.USP18通过调控线粒体功能并抑制线粒体自噬,加剧心肌I/R损伤
5.心肌特异性敲降Parkin会加重USP18-cKO小鼠的急性心肌I/R损伤
为验证USP18是否通过抑制线粒体自噬来加重心肌细胞损伤,在USP18敲低的NRVM中进一步敲低Parkin(或使用抑制剂Mdivi-1)阻断线粒体自噬,结果逆转了USP18沉默对H/R损伤的保护作用(细胞活力下降、LDH释放增加、DNA碎片化、cleaved caspase-3活性升高及线粒体功能障碍)。
与体外实验结果一致,在体内敲低Parkin会加重USP18-cKO小鼠的急性I/R损伤。而且,Parkin敲低显著逆转了USP18敲除对线粒体功能障碍和线粒体自噬失衡的保护作用。此外,USP18-cKO对I/R术后4周不良心脏重构和心功能障碍的保护效应,也在Parkin敲低后被消除。这些体内外实验共同表明,线粒体自噬受损是USP18介导心肌I/R损伤的关键环节。
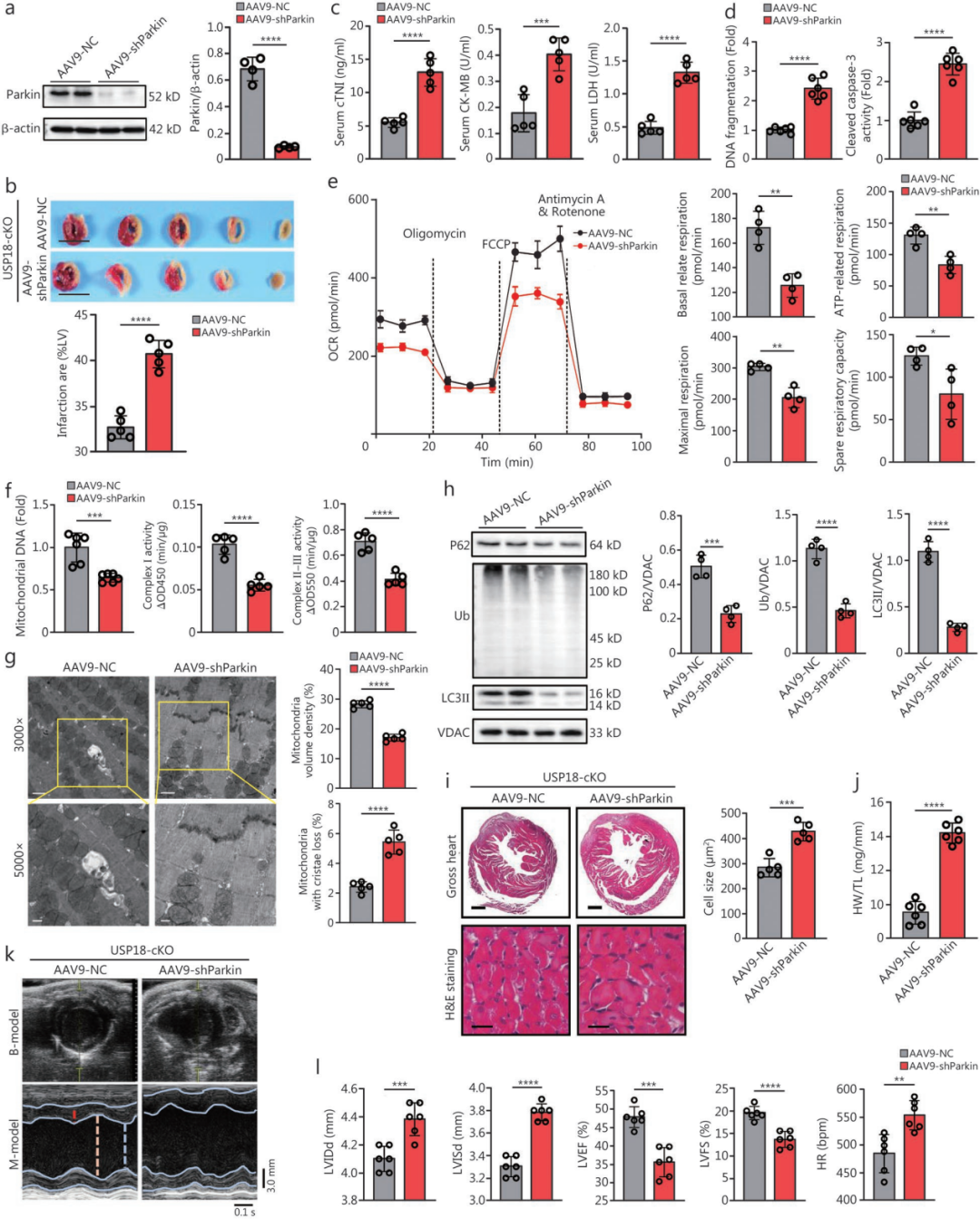
图5.小鼠体内敲低Parkin可抵消USP18缺失的保护作用
6.USP18通过去泛素化上调PTEN-L表达,进而抑制线粒体自噬降解,加剧心肌I/R损伤
为寻找USP18调控线粒体自噬的下游靶点,研究者用免疫共沉淀(Co-IP)联合质谱筛选心肌细胞中与USP18相互作用的蛋白,结果PTEN第一。但后续发现USP18并不调控经典PTEN,而是调控其长亚型PTEN-L:在USP18-cKO心脏和敲低USP18的NRVM中,PTEN-L蛋白水平下降;在USP18过表达(腺病毒或AAV)时,PTEN-L水平升高,而PTEN不变。
Co-IP证实USP18与PTEN-L直接结合,且I/R或H/R处理后两者内源性结合增强。Pull-down实验进一步定位,USP18的1-109 aa和299-368 aa片段与PTEN-L的ATR结构域有强结合。进一步机制表明,USP18通过去泛素化PTEN-L(主要抑制K48链泛素化)来延长其半衰期(放线&菌酮实验证实),从而稳定PTEN-L。
已知pSer65-Ub可招募Parkin至线粒体,而PTEN-L能使pSer65-Ub去磷酸化,从而降低线粒体上的pSer65-Ub水平。本研究发现,在USP18-cKO小鼠心脏及敲低USP18的NRVM中,线粒体pSer65-Ub水平升高;在USP18-OV小鼠或过表达USP18的NRVM中则降低。此外,Co-IP显示PTEN-L与Parkin存在结合,且USP18过表达增强二者结合,敲低则减弱。功能上,PTEN-L抑制Parkin向线粒体转位(亚细胞分离实验),并降低Parkin磷酸化水平(Phos-tag实验)。综上,USP18通过稳定PTEN-L,使其与Parkin结合,进而抑制Parkin的磷酸化和线粒体转位,最终阻断线粒体自噬,加重I/R损伤。
为验证USP18的有害作用是否依赖其去泛素化酶活性,研究者构建了去泛素化失活的USP18突变体(C61A)。结果显示,过表达野生型USP18可加重H/R诱导的线粒体功能障碍和细胞损伤,而C61A突变体无此作用。同样,为验证PTEN-L是否需要其磷酸酶活性,构建了磷酸酶失活的PTEN-L突变体(C297S),发现野生型PTEN-L加重损伤,而C297S无作用。功能回复实验进一步证实了上下游关系:在USP18过表达的NRVM中敲低PTEN-L(siRNA),可减轻损伤,说明USP18的有害作用依赖于PTEN-L;反之,在USP18敲低的NRVM中过表达PTEN-L(腺病毒),损伤仍加重,表明PTEN-L可独立于USP18起有害作用。体内同样用AAV9-shPTEN-L特异性敲低心肌中的PTEN-L,结果减轻了急性I/R损伤、线粒体功能障碍、线粒体自噬失衡以及远期心脏重构和心功能下降;而且,在PTEN-L敲低背景下,USP18过表达不再加重这些病理改变。
综上,USP18通过去泛素化稳定PTEN-L,PTEN-L通过其磷酸酶活性抑制Parkin的转位和磷酸化,从而阻断线粒体自噬,最终加剧心肌I/R损伤。
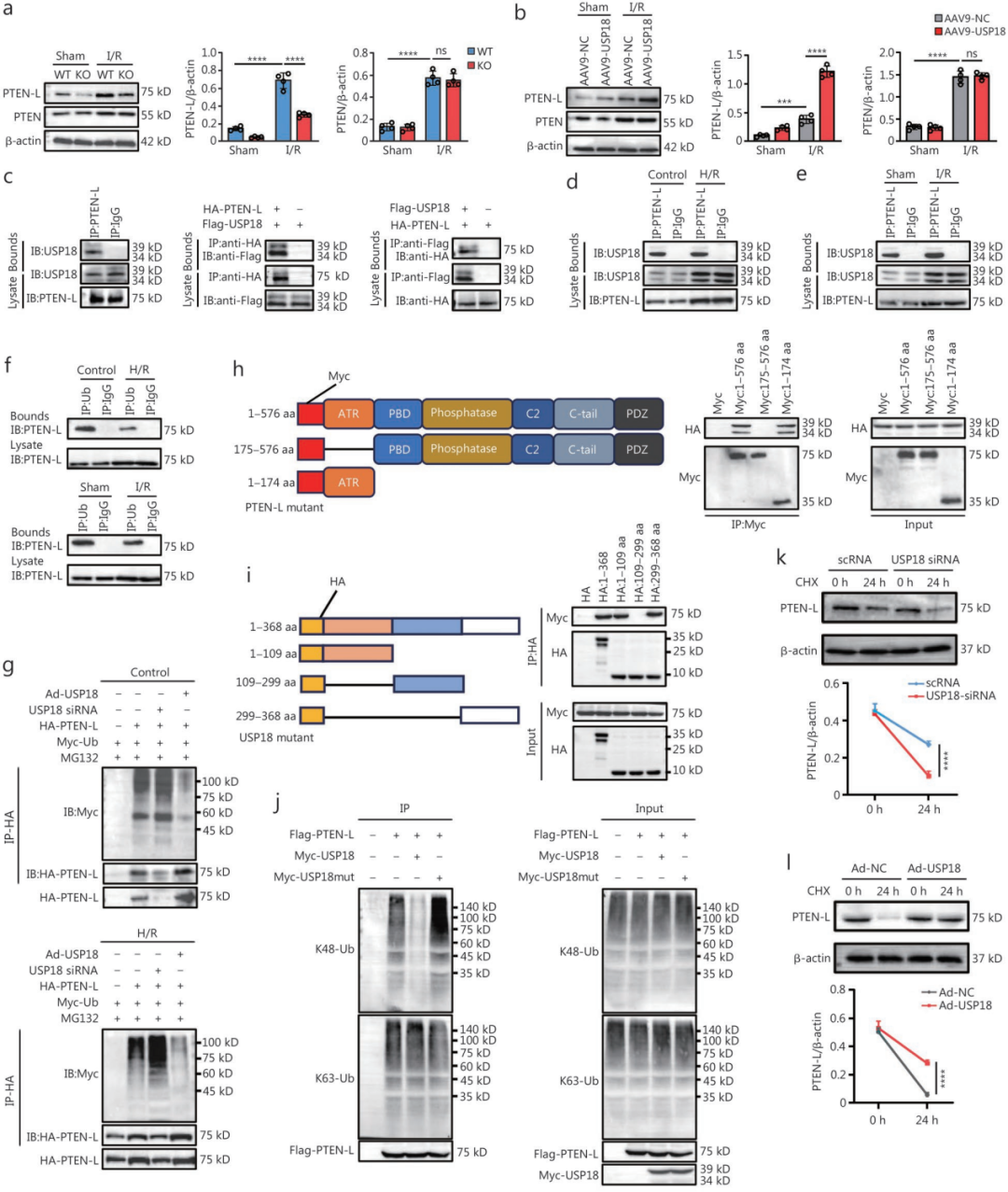
图6.USP18通过去泛素化上调PTEN‑L,从而抑制线粒体自噬降解,加重心脏I/R损伤
7.抗PTEN-L抗体对心脏I/R损伤具有体内保护作用
PTEN-L是一种分泌蛋白,可在血清中检测到。I/R处理后,小鼠血液中PTEN-L水平升高;USP18-OV小鼠更高,USP18-cKO小鼠更低。细胞实验同样显示,过表达USP18使细胞上清中PTEN-L升高,敲低USP18则降低。
PTEN-L通过其ATR结构域介导分泌。当ATR结构域突变后,细胞上清中几乎检测不到PTEN-L,且该突变体不再加重H/R诱导的线粒体功能障碍和细胞损伤。此外,使用抗PTEN-L抗体(I/R术后小鼠隔天注射抗体,共28天),无论体外还是体内,均可减轻H/R或I/R损伤(改善线粒体功能、自噬失衡、心脏重构和心功能)。
临床数据显示,STEMI患者血清PTEN-L水平升高,PCI术后即刻进一步升高,且PTEN-L水平与LVEF、LVFS呈负相关。
综上,I/R上调心脏USP18,USP18稳定PTEN-L,后者抑制Parkin通路及线粒体自噬,并通过旁分泌放大损伤。靶向USP18-PTEN-L轴可恢复自噬、保护心脏。
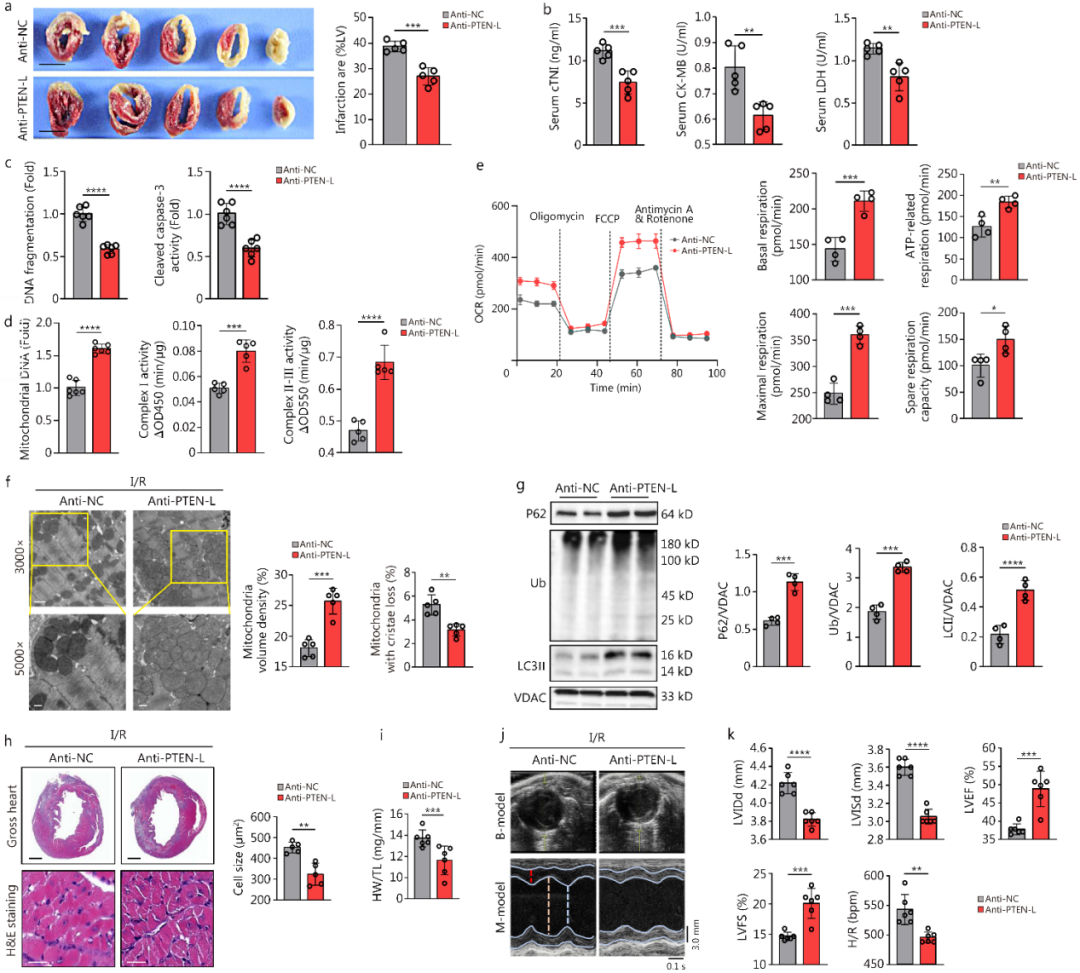
图7.抗PTEN‑L抗体能减轻小鼠体内心脏I/R损伤
总结
综上所述,在心肌I/R损伤过程中,炎症因子促使USP18表达上调,进而减少PTEN-L的泛素化降解,使其水平升高。升高的PTEN-L在细胞质中与Parkin结合,阻碍Parkin向线粒体转位,从而抑制线粒体自噬的降解功能,最终加剧心脏损伤。USP18被鉴定为一种在心肌细胞中高表达、与炎症相关的有害因子,并可作为心肌I/R损伤富有前景的新型治疗靶点,靶向USP18/PTEN-L信号通路能够有效改善心肌I/R损伤。
既往研究(Ying X, et al. Hypertension. 2016)曾指出USP18在压力负荷引起的心脏重构中具有保护作用(缓解心肌肥厚和纤维化),然而本研究发现,在心肌I/R损伤中USP18反而是有害的。具体来说,过表达USP18会加剧线粒体功能障碍、加重心肌I/R损伤,并使心脏重构和心功能进一步恶化。造成这种差异的关键在于两种模型所涉及的病理生理背景和细胞应激类型不同:压力负荷模型(如主动脉缩窄)主要激活肥厚信号和纤维化重构,而I/R损伤则以急性氧化应激、线粒体功能障碍和线粒体自噬激活为特征。这些结果提示,USP18的作用具有明显的刺激特异性和通路依赖性,即在某些慢性心脏重构的背景下它可能发挥保护作用,但在急性缺血性损伤中,由于它损害了线粒体稳态,反而成为有害因素。因此,将USP18作为治疗靶点时,必须根据具体的疾病背景谨慎考量。
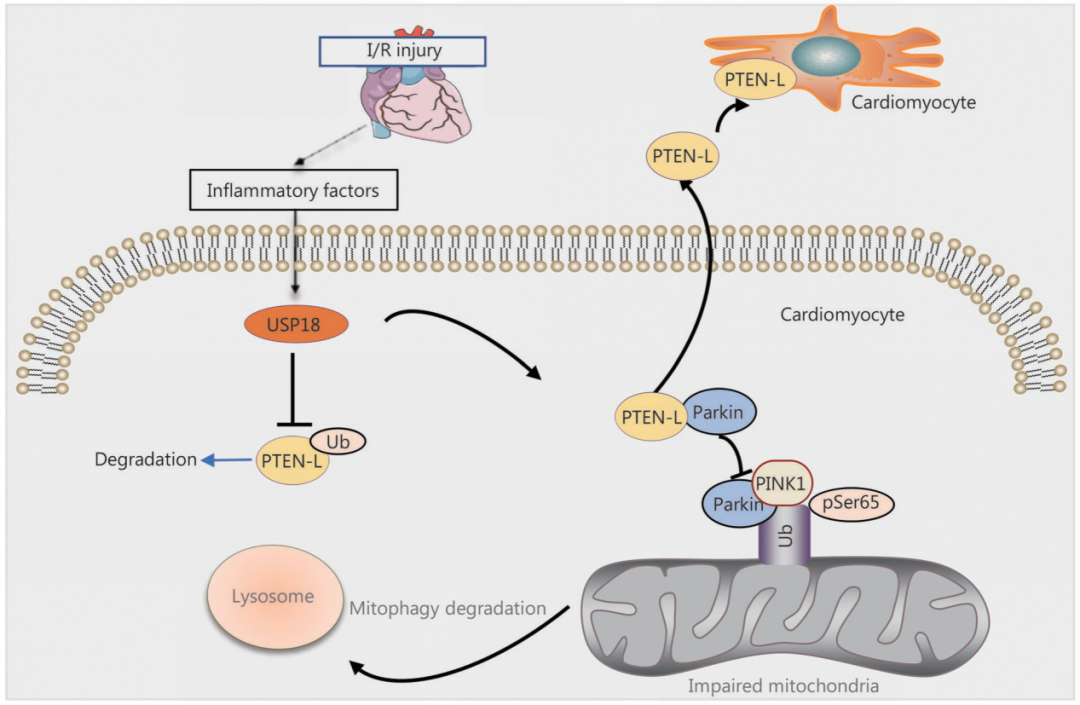
USP18-PTEN-L轴抑制线粒体自噬加重心肌I/R损伤模式图
本文使用的病毒产品,列表如下:
上述病毒产品我司均可提供,欢迎咨询!
如有相关需求,或了解更多产品服务,欢迎咨询我们!点击进入店铺,查看更多产品及服务